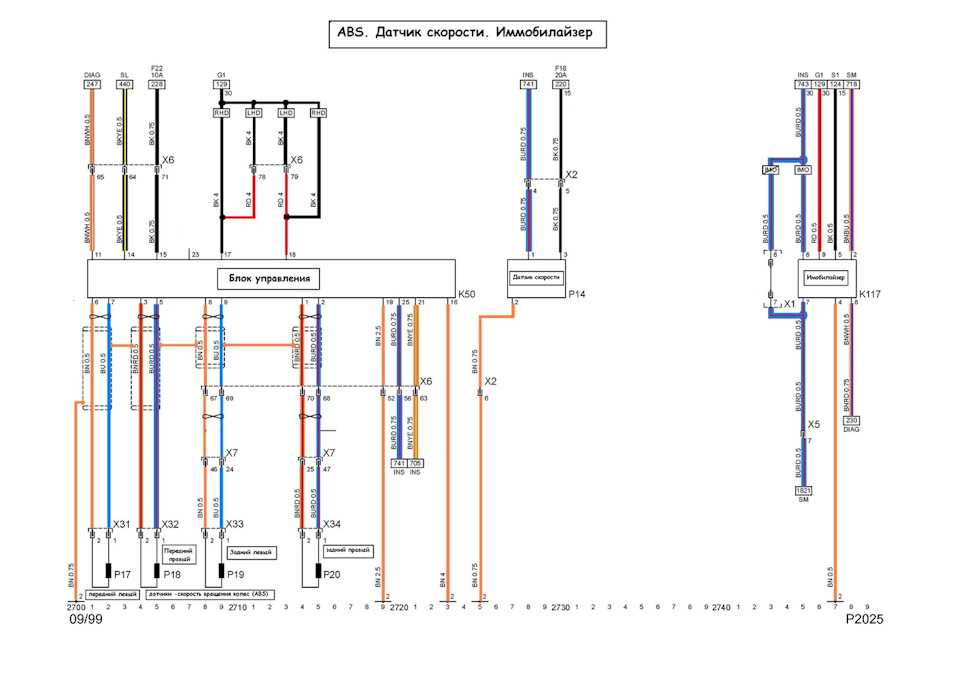
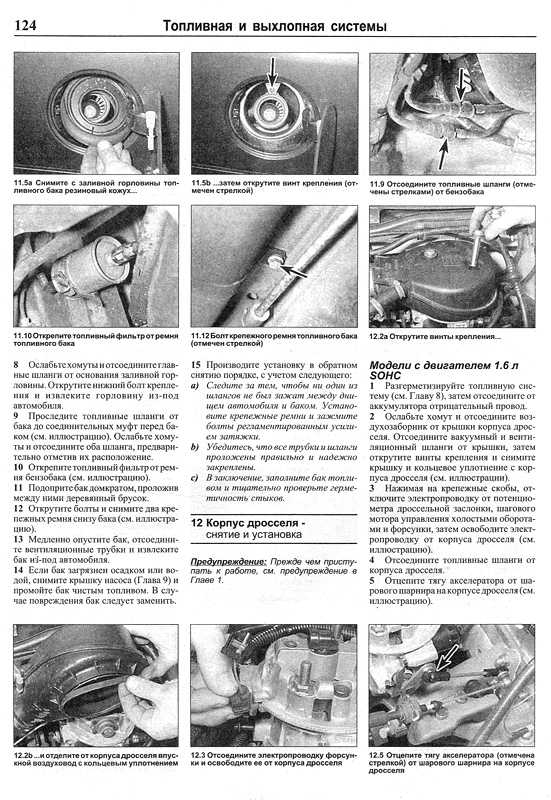
Собрание руководств по ремонту и обслуживанию всех моделей Опель своими руками. Статьи, фото и видео инструкции по ремонту двигателя, кузова и систем авто.


.

Ремонт и обслуживание автомобилей opel zafira своими руками Ремонт Опель Зафира 1.8 Своими Руками